
Research
Our lab works on both the application of cryo-EM for biomolecular structure determination
and also the development of computational tools for cryo-EM.
We have a focus on the determination of
structures of amyloid fibrils in collaboration with our colleagues at the
ICS-6 in Jülich and the IPB in Düsseldorf.
Amyloid fibrils play a significant role in many (but not only in) neurodegenerative diseases, such as Alzheimers or Parkinsons disease.
Cryo-EM Structure Projects
Amyloid-β(1-42) fibril structure
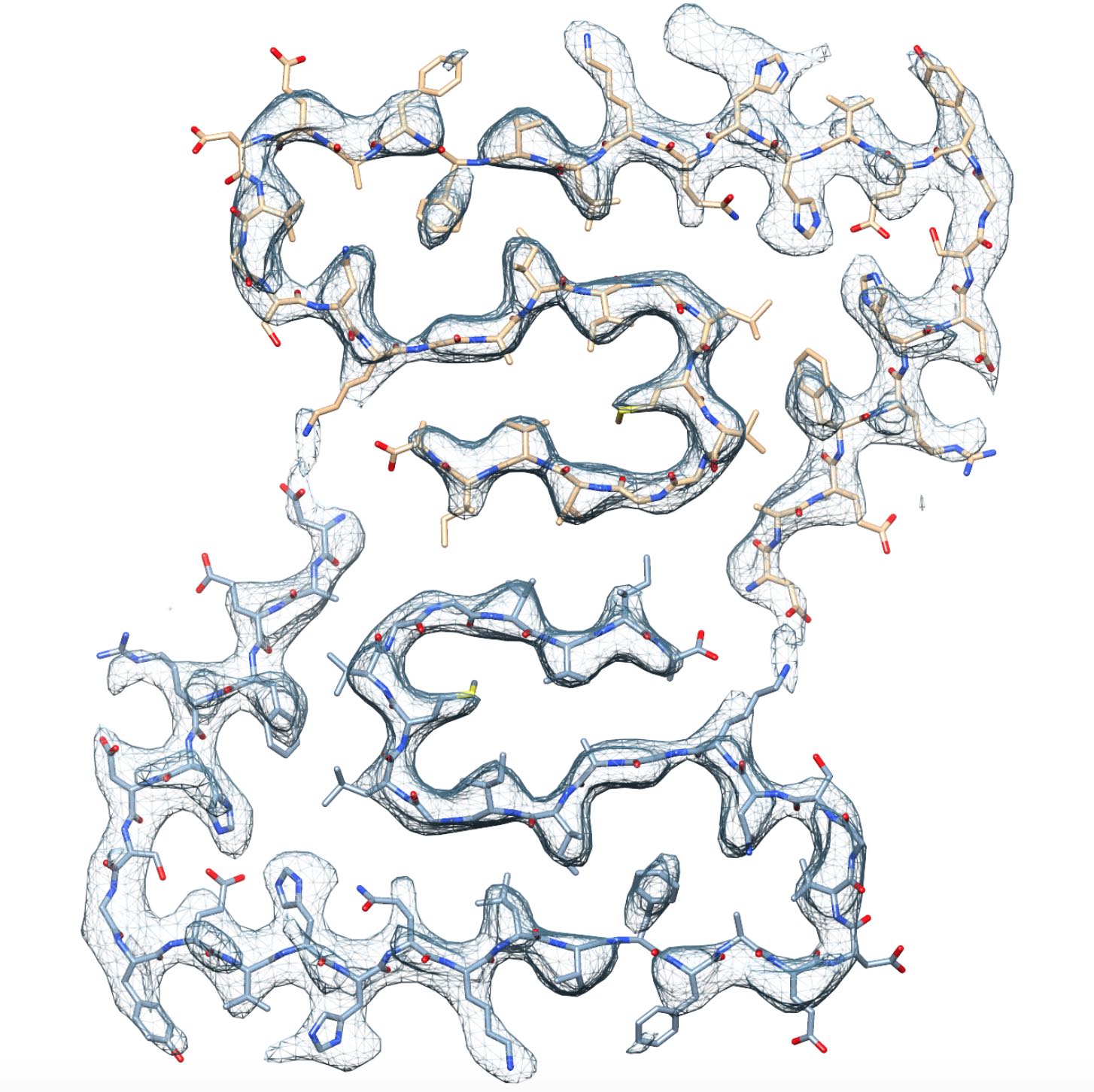 Fibrillar aggregates of the amyloid-β protein (Aβ) are the main component of
the senile plaques found in brains of Alzheimer’s disease patients.
We have determined the structure of an Aβ(1-42) fibril composed of two
intertwined protofilaments determined by cryo-EM to 4.0 Å resolution,
complemented by solid-state nuclear magnetic resonance (NMR) experiments.
The backbone of all 42 residues and nearly all sidechains are well resolved
in the EM density map, including the entire N-terminus, which is part of
the cross-β structure resulting in an overall "LS"-shaped topology of
individual subunits. The dimer interface protects the hydrophobic C-termini
from the solvent. The unique staggering of the non-planar subunits
results in markedly different fibril ends, termed "groove" and "ridge",
leading to different binding pathways on both fibril ends, which has
implications for fibril growth.
Fibrillar aggregates of the amyloid-β protein (Aβ) are the main component of
the senile plaques found in brains of Alzheimer’s disease patients.
We have determined the structure of an Aβ(1-42) fibril composed of two
intertwined protofilaments determined by cryo-EM to 4.0 Å resolution,
complemented by solid-state nuclear magnetic resonance (NMR) experiments.
The backbone of all 42 residues and nearly all sidechains are well resolved
in the EM density map, including the entire N-terminus, which is part of
the cross-β structure resulting in an overall "LS"-shaped topology of
individual subunits. The dimer interface protects the hydrophobic C-termini
from the solvent. The unique staggering of the non-planar subunits
results in markedly different fibril ends, termed "groove" and "ridge",
leading to different binding pathways on both fibril ends, which has
implications for fibril growth.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, Jörg Labahn, R.B.G. Ravelli, M. Tusche, C. Lopez- Iglesias, W. Hoyer, H. Heise, D. Willbold, G.F. Schröder. Science 358:116–119 (2017)
- (Protein Databank) PDB ID: 5OQV
PI3K-SH3 fibril structure
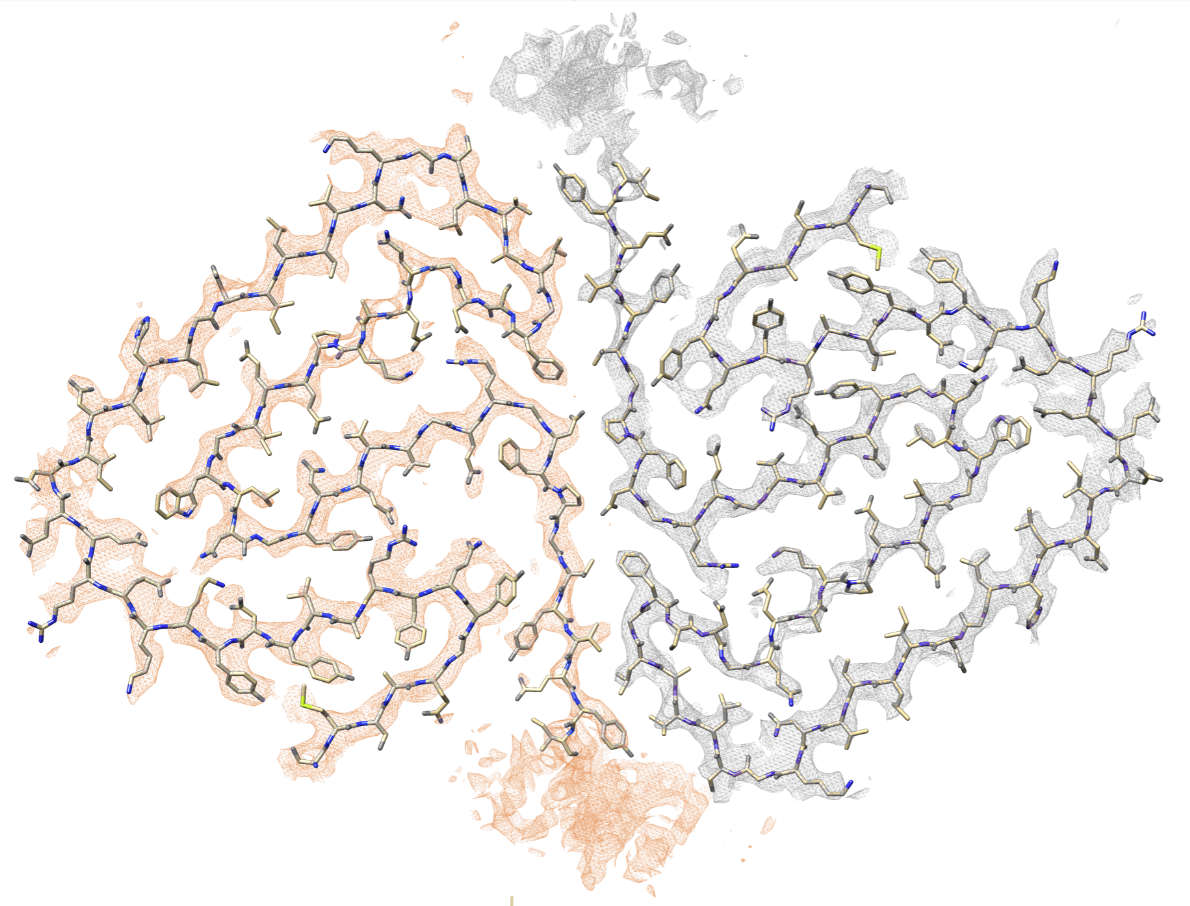 The Src-homology 3 domain of phosphatidyl-inositol-3-kinase (PI3K-SH3) is a model
amyloid system that plays a pivotal role in our
basic understanding of protein misfolding and aggregation. We have determined the atomic
model of the PI3K-SH3 amyloid fibril with a resolution of 3.4 Å by
cryo-EM. The fibril is composed of two intertwined protofilaments
that create an interface spanning 13 residues from each monomer. The model
comprises residues 1–77 out of 86 amino acids in total, with the missing residues
located in the highly flexible C-terminus. The fibril structure allows us to rationalise the
effects of chemically conservative point mutations as well as of the previously reported
sequence perturbations on PI3K-SH3 fibril formation and growth.
The Src-homology 3 domain of phosphatidyl-inositol-3-kinase (PI3K-SH3) is a model
amyloid system that plays a pivotal role in our
basic understanding of protein misfolding and aggregation. We have determined the atomic
model of the PI3K-SH3 amyloid fibril with a resolution of 3.4 Å by
cryo-EM. The fibril is composed of two intertwined protofilaments
that create an interface spanning 13 residues from each monomer. The model
comprises residues 1–77 out of 86 amino acids in total, with the missing residues
located in the highly flexible C-terminus. The fibril structure allows us to rationalise the
effects of chemically conservative point mutations as well as of the previously reported
sequence perturbations on PI3K-SH3 fibril formation and growth.
- C. Röder, N. Vettore, L. N. Mangels, L. Gremer, R.B.G. Ravelli, D. Willbold, W. Hoyer, A.K. Buell, G.F. Schröder Atomic structure of PI3-kinase SH3 amyloid fibrils by cryo-electron microscopy Nat. Commun. (2019)
- (Protein Databank) PDB ID: 6R4R
Structures determined in collaborations:
Actin Filaments
Work on actin filaments was done in collaboration with the groups of Ed Egelman and Vitold Galkin.-
C. Risi, B. Belknap,
E. Forgacs-Lonart,
S. P. Harris,
G. F. Schröder, H. D. White, V. E. Galkin
N-Terminal Domains of Cardiac Myosin Binding Protein C Cooperatively Activate the Thin Filament
Structure (2018) 26:1-8 -
C. Risi, J. Eisner, B. Belknap, D. H. Heeley, H. D. White, G. F. Schröder, V. E. Galkin.
Ca2+-induced Movement of Tropomyosin on Native Cardiac Thin Filaments Revealed by Cryo Electron Microscopy
PNAS (2017) 114:6782-6787 -
T. Braun, M. Vos, N. Kalisman, N. E. Sherman, R. Rachel, R. Wirth, G.F. Schröder, and E. Egelman.
Archaeal Flagellin Combines a Bacterial Type IV Pilin Domain with an Immunoglobulin-like Domain
PNAS (2016) 113(37):10352-7 -
T. Braun,
A. Orlova,
K. Valegård,
A.-C. Lindås,
G.F. Schröder, and
E. H. Egelman.
An Archaeal Actin from a Hyperthermophile Forms a Single-Stranded Filament
PNAS (2015), 112(30): 9340-9345. -
V.E. Galkin, A. Orlova, M.R. Vos, G.F. Schröder, and E.H. Egelman.
Near-Atomic Resolution for One State of F-Actin
Structure (2015) 23(1): 173-182
V.E. Galkin, A. Orlova, D. Kudryashov, A. Solodukhin, E. Reisler, G.F. Schröder, and E.H. Egelman.
Remodeling of Actin Filaments by ADF/Cofilin Proteins.
-
V.E. Galkin, A. Orlova, G.F. Schröder, E.H. Egelman.
Structural Polymorphism in F-actin.
Nat Struct Mol Biol (2010) 17:1318-1323. [PDF]
PNAS (2011) 108(51):20568-20572 . [PDF]
Ribosome Structures and Dynamics
Work on ribosome structures was done in collaboration with the group of Holger Stark.- N. Fischer, P. Neumann, L. V. Bock, C. Maracci, Z. Wang, A. Paleskava, A. L. Konevega, G. F. Schröder, H. Grubmüller, R. Ficner, M. V. Rodnina, and H. Stark. The pathway to GTPase activation of elongation factor SelB on the ribosome Nature (2016) 540:80-85
- L.V. Bock, C. Blau, G.F. Schröder, I.I. Davydov, N. Fischer, H. Stark, M.V. Rodnina, A.C. Vaiana, and H. Grubmüller. Energy barriers and driving forces in tRNA translocation through the ribosome Nat Struct Mol Biol (2013) 20:1390–1396
Chaperones (GroEL and CCT)
The work on chaperones was done mostly together with the group of Wah Chiu.- D.-H. Chen, D. Madan, J. Weaver, Z. Lin, G.F. Schröder, W. Chiu, H.S. Rye. Visualizing GroEL/ES in the Act of Encapsulating a Folding Protein Cell (2013) 153:1354–1365. [PDF]
- N. Kalisman, G.F. Schröder, M. Levitt. The Crystal Structures of the Eukaryotic Chaperonin CCT Reveal its Functional Partitioning Structure (2013) 21:540–549. [PDF]
- Y. Cong, G.F. Schröder, A.S. Meyer, J. Jakana, B. Ma, M.T. Dougherty, M.F. Schmid, S. Reissmann, M. Levitt, S.L. Ludtke, J. Frydman, and W. Chiu. Symmetry-Free Cryo-EM Structures of the Chaperonin TRiC Along its ATPase-Driven Conformational Cycle. EMBO J (2011) 31:720-730. [PDF]
-
J. Zhang, M.L. Baker, G.F. Schröder, N.R. Douglas,
S. Reissmann, J. Jakana, M. Dougherty, C.J. Fu, M. Levitt, S.J. Ludtke,
J. Frydman, and W. Chiu.
Mechanism of folding chamber closure in a group II chaperonin.
Nature (2010) 463:379-383. [PDF]
Cryo-EM Computational Methods
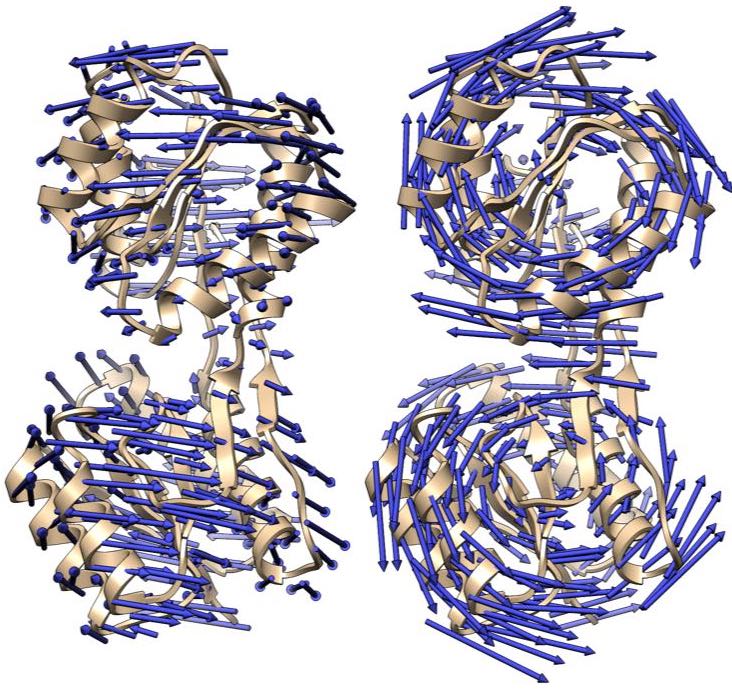
Analyzing molecular motions from cryo-EM Data
Single-particle cryo-EM images provide information on the conformational distribution of biomolecules,, and therefore on conformational motions of these molecules. We are developing the principal motion analysis (PMA) method to determine these conformational motions.Cross-validation in structure refinement
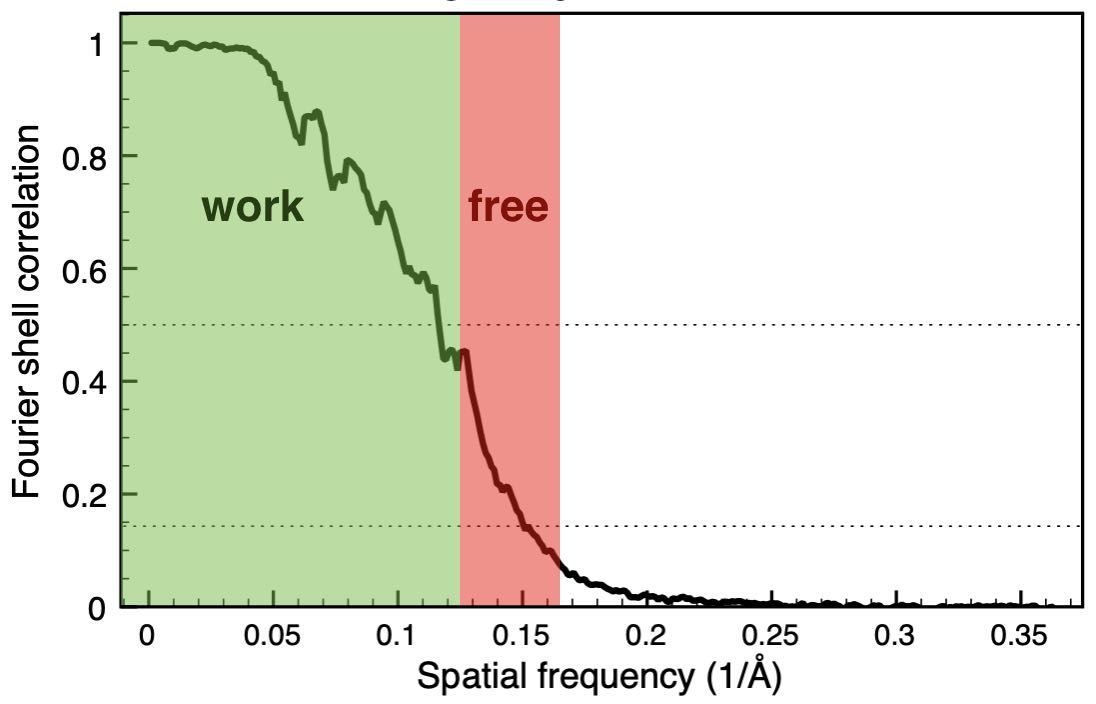 We have developed a cross-validation approach for
real-space refinement against cryo-EM density maps in analogy to cross-validation
typically used in crystallography. Our approach is able to detect overfitting
and allows for optimizing the choice of restraints used in the refinement.
We treat a high-resolution Fourier shell as a the test set, that is not used
for the refinement, but only for validation.
Because cross-validation
requires splitting the dataset into at least two independent sets, we further
present an approach to quantify correlations between the structure factor sets.
This analysis is also helpful for other cross-validation applications, such
as refinements against diffraction data or 3D reconstructions of cryo-EM
density maps.
We have developed a cross-validation approach for
real-space refinement against cryo-EM density maps in analogy to cross-validation
typically used in crystallography. Our approach is able to detect overfitting
and allows for optimizing the choice of restraints used in the refinement.
We treat a high-resolution Fourier shell as a the test set, that is not used
for the refinement, but only for validation.
Because cross-validation
requires splitting the dataset into at least two independent sets, we further
present an approach to quantify correlations between the structure factor sets.
This analysis is also helpful for other cross-validation applications, such
as refinements against diffraction data or 3D reconstructions of cryo-EM
density maps.
-
B. Falkner and G.F. Schröder.
Cross-validation in cryo-electron microscopy based structural modeling
PNAS (2013) 110(22):8930-8935. [PDF]
Clustering of helical polymer images
 Helical protein polymers are often dynamic and complex assemblies, with many
conformations and flexible domains possible within the helical assembly.
During cryo-EM reconstruction, classification of the image data
into homogeneous subsets is a critical step for achieving high resolution,
resolving different conformations and elucidating functional mechanisms.
Hence, methods aimed at improving the homogeneity of these datasets are becoming
increasingly important.
We have developed an algorithm that uses
results from 2D image classification to sort 2D classes into groups of similar
helical polymers.
Helical protein polymers are often dynamic and complex assemblies, with many
conformations and flexible domains possible within the helical assembly.
During cryo-EM reconstruction, classification of the image data
into homogeneous subsets is a critical step for achieving high resolution,
resolving different conformations and elucidating functional mechanisms.
Hence, methods aimed at improving the homogeneity of these datasets are becoming
increasingly important.
We have developed an algorithm that uses
results from 2D image classification to sort 2D classes into groups of similar
helical polymers.
-
K. R. Pothula,
D. Smyrnova,
G. F. Schröder.
Clustering cryo-EM images of helical protein polymers for helical reconstructions
Ultramicroscopy (2018) 203:132-138
Density Sharpening: VISDEM
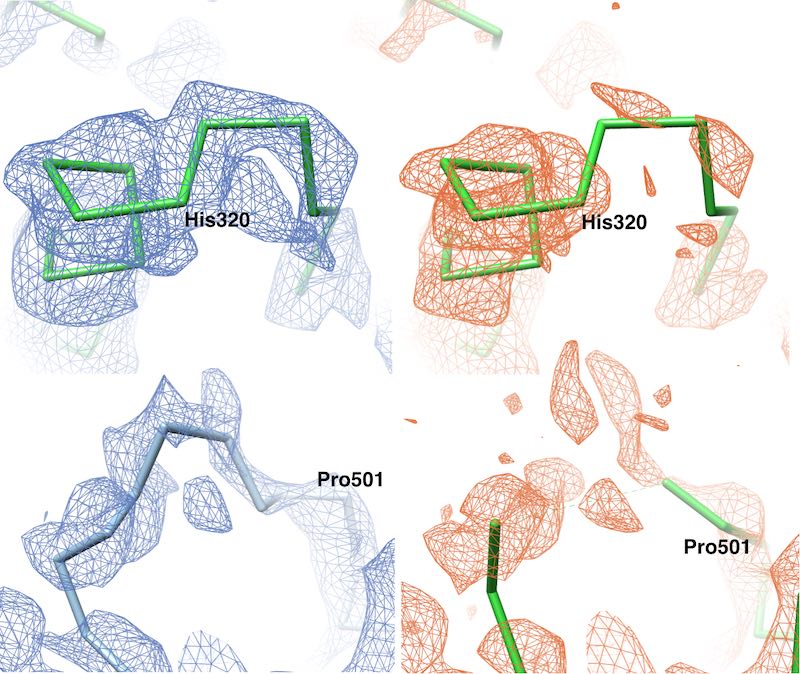 We have developed a method to improve the visualisation of density maps by using general
statistical information about proteins for the sharpening process. In particular, the packing
density of atoms is highly similar between different proteins, which allows for building a
pseudo-atomic model to approximate the true mass distribution. From this model the radial
structure factor and density value histogram are estimated and applied as constraints to
the 3D reconstruction in reciprocal- and real-space, respectively. Interestingly, similar
improvements are obtained when using the correct radial structure factor and density value
histogram from a crystal structure. Thus, the estimated pseudo-atomic model yields a
suffciently accurate mass distribution to optimally sharpen a density map.
We have developed a method to improve the visualisation of density maps by using general
statistical information about proteins for the sharpening process. In particular, the packing
density of atoms is highly similar between different proteins, which allows for building a
pseudo-atomic model to approximate the true mass distribution. From this model the radial
structure factor and density value histogram are estimated and applied as constraints to
the 3D reconstruction in reciprocal- and real-space, respectively. Interestingly, similar
improvements are obtained when using the correct radial structure factor and density value
histogram from a crystal structure. Thus, the estimated pseudo-atomic model yields a
suffciently accurate mass distribution to optimally sharpen a density map.
-
M. Spiegel, A.K. Duraisamy, and G.F. Schröder.
Improving the Visualisation of Cryo-EM Density Reconstructions
J.Struct.Biol. (2015), 191(2):207-213
Real-space model refinement with DireX
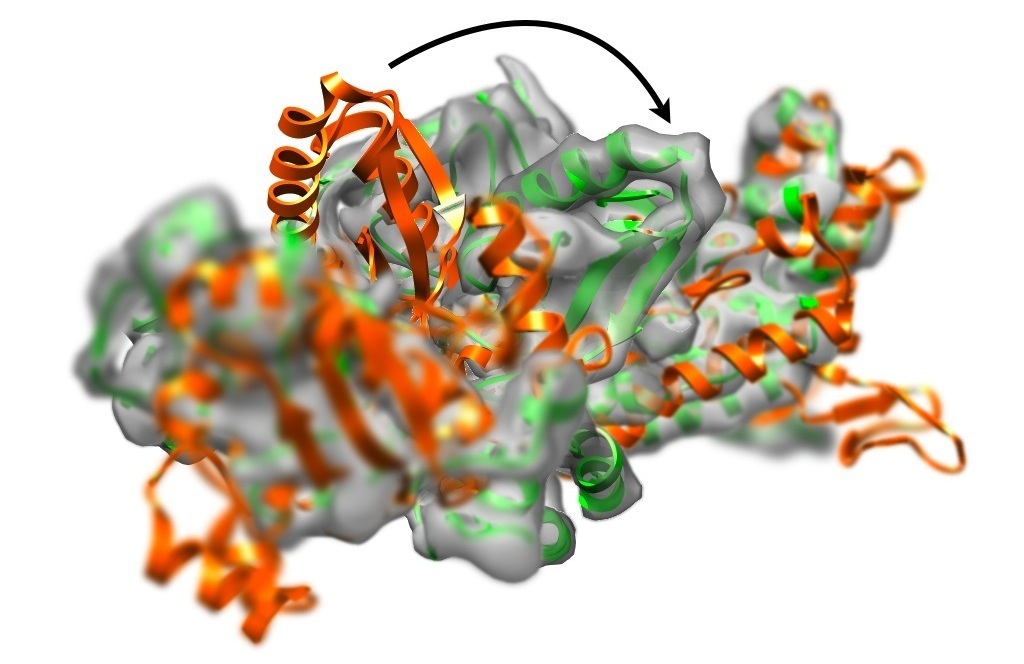 Single-particle cryo-electron microscopy (CryoEM) is able to determine
three-dimensional density distributions (density maps) of large macromolecules.
The resolution ranges from (in few cases) higher than 4 Å to often well below 10 Å.
In case high-resolution structures (e.g. from crystallography) is available
the goal is to refine the structure to fit into the low-resolution CryoEM density map.
For this purpose we have developed the real-space refinement program DireX.
Single-particle cryo-electron microscopy (CryoEM) is able to determine
three-dimensional density distributions (density maps) of large macromolecules.
The resolution ranges from (in few cases) higher than 4 Å to often well below 10 Å.
In case high-resolution structures (e.g. from crystallography) is available
the goal is to refine the structure to fit into the low-resolution CryoEM density map.
For this purpose we have developed the real-space refinement program DireX.
Read more about DireX and watch movies of the flexible fitting...
Deformable Elastic Network (DEN)
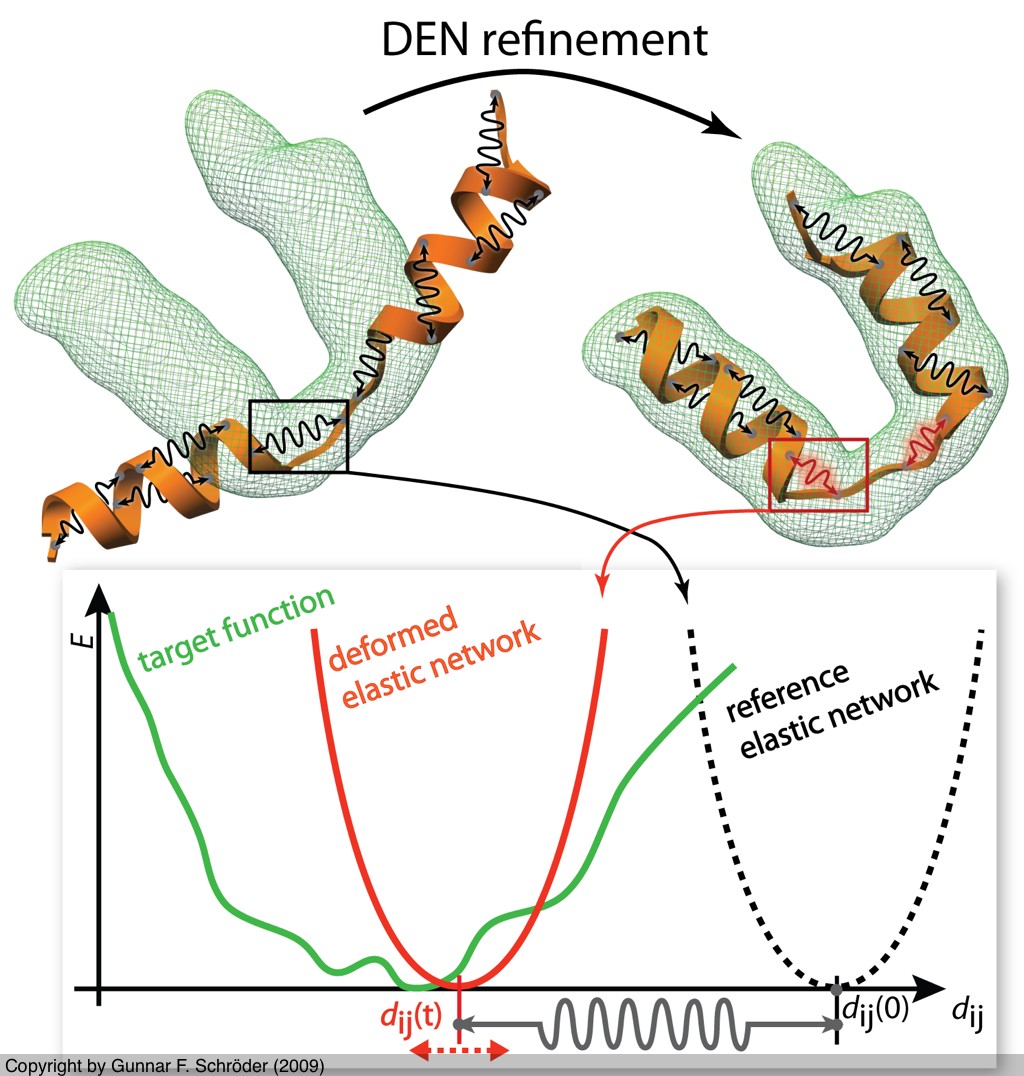 We developed the Deformable Elastic Network approach to combine
prior structural knowledge with low-resolution data.
The idea is to adapt an elastic network potential (defined on a reference structure)
in a way that
deforms only those degrees of freedom that need to be deformed to fit the data
but not more. This approach significantly reduces overfitting, which is usually
encountered in low-resolution refinement.
We developed the Deformable Elastic Network approach to combine
prior structural knowledge with low-resolution data.
The idea is to adapt an elastic network potential (defined on a reference structure)
in a way that
deforms only those degrees of freedom that need to be deformed to fit the data
but not more. This approach significantly reduces overfitting, which is usually
encountered in low-resolution refinement.
- G.F. Schröder, A.T. Brunger, and M. Levitt. Structure (2007) 15:1630-1641. [PDF]
Structural Interpretation of FRET Experiments
Modeling based on multiple FRET Experiments
 Single-molecule Förster Resonance Energy Transfer (smFRET) measurements use the non-radiative transfer of energy from a donor to an acceptor dye. By monitoring the relative fluorescence intensities from the two dyes, the energy transfer efficiency can be evaluated for individual molecules. The transfer efficiency is commonly employed to determine the interdye distance. This method has emerged as a powerful biophysical tool
to measure the distance between two labeled sites within one macromolecule or between two labeled macromolecules.
FRET measurements are most sensitive in the 10-100 Å distance range.
With several of such measured distances
it becomes possible to build a three-dimensional model and to describe conformational transitions. We developed
methods for this purpose and applied them to different kinds of
systems from proteins to DNA and RNA.
Single-molecule Förster Resonance Energy Transfer (smFRET) measurements use the non-radiative transfer of energy from a donor to an acceptor dye. By monitoring the relative fluorescence intensities from the two dyes, the energy transfer efficiency can be evaluated for individual molecules. The transfer efficiency is commonly employed to determine the interdye distance. This method has emerged as a powerful biophysical tool
to measure the distance between two labeled sites within one macromolecule or between two labeled macromolecules.
FRET measurements are most sensitive in the 10-100 Å distance range.
With several of such measured distances
it becomes possible to build a three-dimensional model and to describe conformational transitions. We developed
methods for this purpose and applied them to different kinds of
systems from proteins to DNA and RNA.
- A. K. Wozniak, G.F. Schröder, H. Grubmüller, C.A.M. Seidel, F. Oesterhelt. PNAS (2008) 105(47):18337-18342. [PDF]
- A. Wozniak, S. Nottrott, E. Kühn-Hölsken, G. F. Schröder, H. Grubmüller, R. Lührmann, A. M. Seidel and F. Oesterhelt. RNA (2005) 11:1545-1554. [PDF]
- M. Margittai, J. Widengren, E. Schweinberger, G.F. Schröder, S. Felekyan, E. Haustein, M. König, D. Fasshauer, H. Grubmüller, R. Jahn, and C. A. M. Seidel. PNAS (2003) 100(26):15516-15521. [PDF]
- G.F. Schröder and H. Grubmüller. J. Chem. Phys. (2003) 119(18):9920-9924. [PDF]
- G.F. Schröder and H. Grubmüller. Comp. Phys. Comm. (2003) 158:150-157. [PDF]
Simulation of Fluorescence Anisotropy Experiments
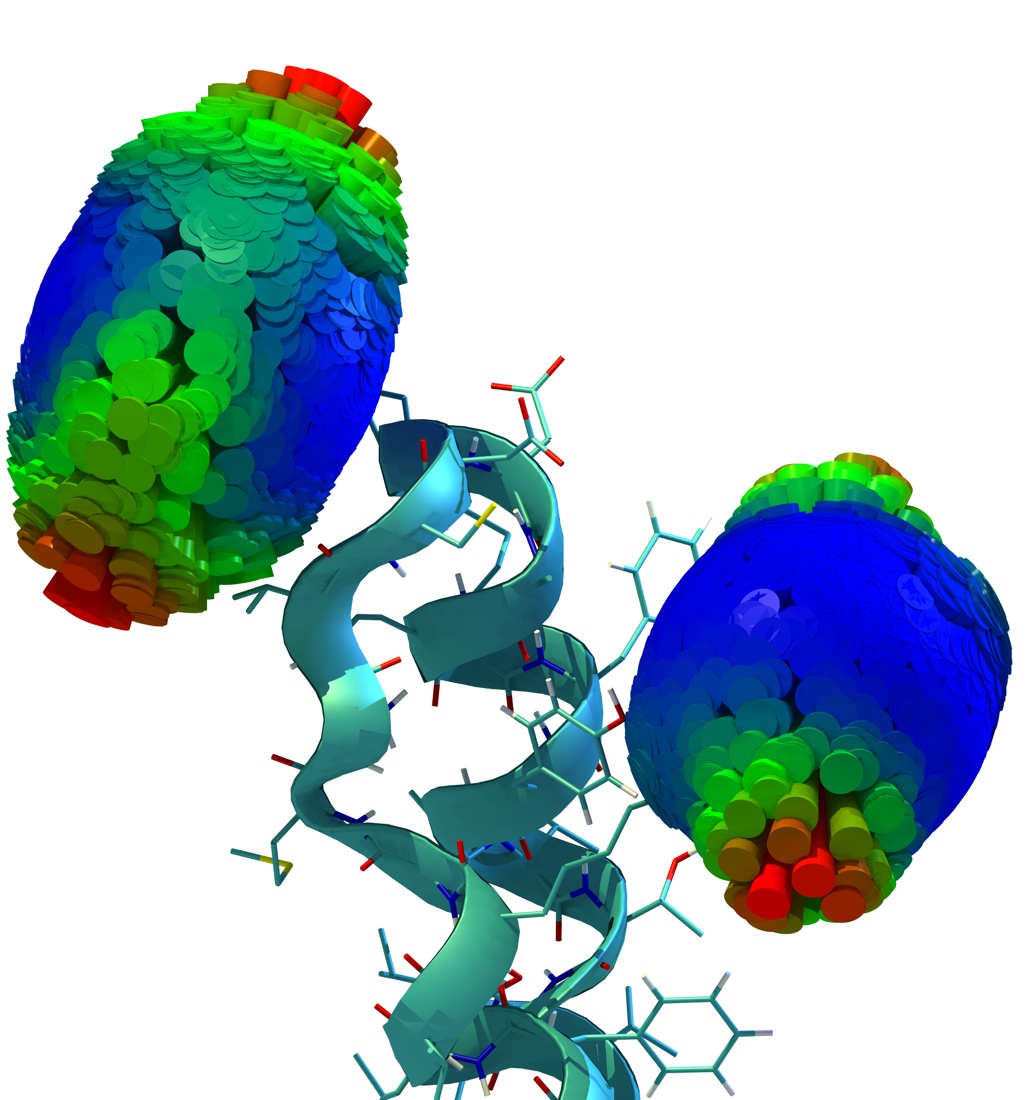 Time-resolved fluorescence anisotropy decay experiments on a protein-attached dye can probe local protein
dynamics and steric restrictions, but are difficult to interpret at the structural level. Aiming at an atomistic description, we have
carried out molecular dynamics simulations of such experiments. Our simulations describe an Alexa488 fluorescent dye maleimide
derivative covalently attached via a single cysteine to the AB-loop of bacteriorhodopsin. Fluorescence anisotropy decay
curves obtained from the simulations agree well with the measured ones.
Furthermore, our simulations
allowed us to test the usual and inevitable assumption underlying these types of spectroscopic measurements that the attached
dye probe does not severely perturb the protein dynamics. For the case at hand, by comparison with a simulation of the dye-free
protein, the perturbation was quantified and found to be small.
Time-resolved fluorescence anisotropy decay experiments on a protein-attached dye can probe local protein
dynamics and steric restrictions, but are difficult to interpret at the structural level. Aiming at an atomistic description, we have
carried out molecular dynamics simulations of such experiments. Our simulations describe an Alexa488 fluorescent dye maleimide
derivative covalently attached via a single cysteine to the AB-loop of bacteriorhodopsin. Fluorescence anisotropy decay
curves obtained from the simulations agree well with the measured ones.
Furthermore, our simulations
allowed us to test the usual and inevitable assumption underlying these types of spectroscopic measurements that the attached
dye probe does not severely perturb the protein dynamics. For the case at hand, by comparison with a simulation of the dye-free
protein, the perturbation was quantified and found to be small.
- G. F. Schröder, U. Alexiev, and H. Grubmüller. Biophys. J. (2005) 89:3757-3770. [PDF]