
DireX - Tutorial
Kinked Helix at Low-resolution
This tutorial shows a very basic example of the usage of DireX to flexibly fit a protein structure into a low-resolution density map. The fitting of this small helix is done in just a few seconds, which allows to conveniently test the effect of the various input parameters.
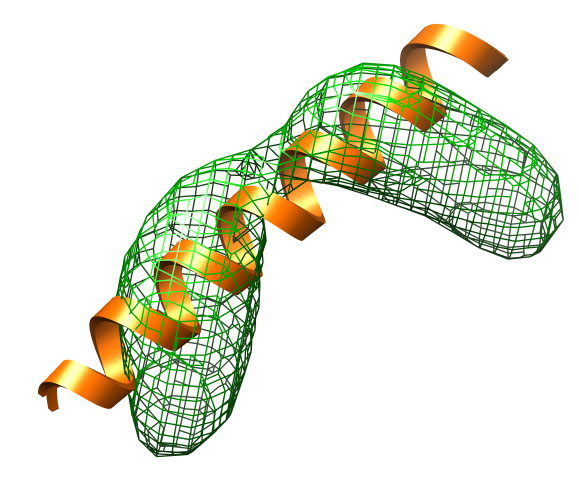 A perfect α-helix (extended-helix.pdb) built with Chimera is
used as the starting structure. This starting structure
serves in this case also as the DEN reference structure
(see Introduction to DEN Method).
The target structure (target.pdb) is a kinked helix that
has a root-mean-square deviation (RMSD) of about 4.7 Å.
We first create a 10 Å density map based on this (kinked) target
structure map with DireX.
The program call can be found in the run.sh bash script:
A perfect α-helix (extended-helix.pdb) built with Chimera is
used as the starting structure. This starting structure
serves in this case also as the DEN reference structure
(see Introduction to DEN Method).
The target structure (target.pdb) is a kinked helix that
has a root-mean-square deviation (RMSD) of about 4.7 Å.
We first create a 10 Å density map based on this (kinked) target
structure map with DireX.
The program call can be found in the run.sh bash script:
The goal is now to fit the straight extended helix into the kinked density map to obtain a fitted structure that gets as close as possible to the target structure.
The actual structure refinement is then performed with (see also the run.sh script):
IMPORTANT: DireX determines the bond lengths and angles directly from the PDB file and it does not optimize those values. It is therefore recommended that you use a (energy minimized) structure with correct bond lengths and angles.
Also, note that only lines starting with "ATOM" and "HETATM" are read, everything else is ignored.
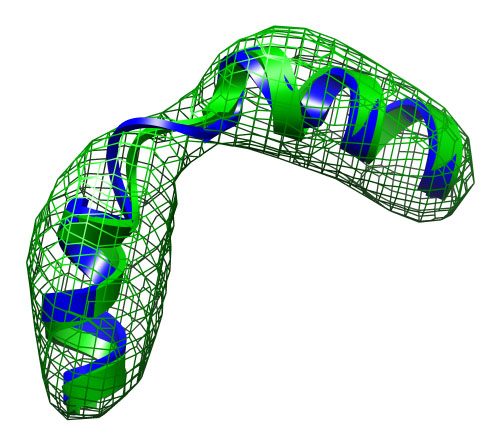 The file current.pdb holds the current coordinates and is updated after
each iteration. After the refinement is finished it therefore
contains the final fitted structure.
The file current.pdb holds the current coordinates and is updated after
each iteration. After the refinement is finished it therefore
contains the final fitted structure.
Analysis
The final fitted structure has an RMSD (Ca-atoms) of 0.9 Å to the target structure. The file mapcc.dat contains the correlation between the model map and the target density map. This file can be viewed e.g. with the plot program xmgr (or grace).
Optimization
An important part of finding the optimal parameters is to balance the fitting to the density map and at the same time obtain a reasonable structure in terms of local geometry, ramachandran statistics, and secondary structure. The DEN restraints are the key element in DireX to control this balance. In the next few steps, we will play with some parameters that define the DEN restraints:
den_no:
The number of restraints should be at least equal to the number of atoms, but typically a better choice would be factor of two. In this case we therefore use 412.
den_gamma:
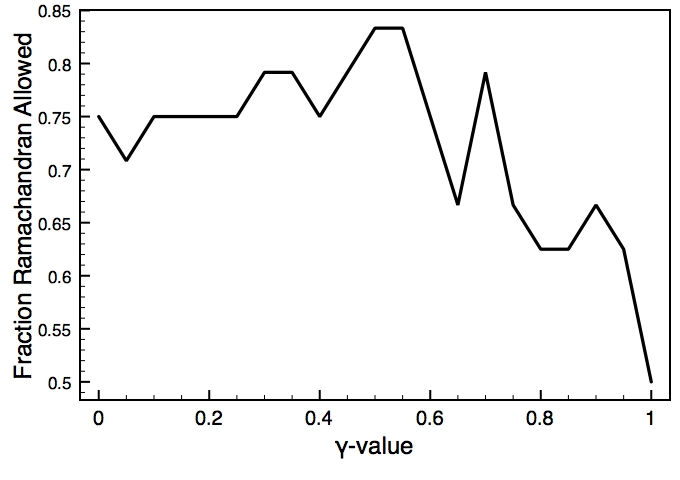
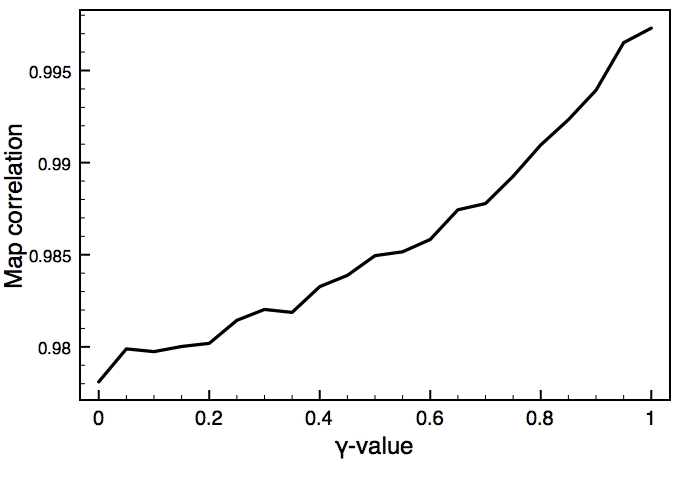
This is one of the key parameters in the DEN approach. In the present case, the DEN reference model is the same as the starting model (extended-helix.pdb). A small γ-value will keep the structure close to this reference model, and a large γ-value will allow larger deformations of the network and therefore should result in higher map correlations. There is usually an optimum choice of den_gamma that best balances reference information and density fit. In principle, this optimum value should be determined by cross-validation (this is not yet part of the tutorials). Since we know the correct target structure for this case, we will compute the RMSD (root mean square deviation) to the target structure to assess the effect of the γ-value. The three plots on the right show the RMSD, the fraction of residues that are within the allowed region of the Ramchandran plot, and the density map correlation. From the RMSD plot, the optimal γ-value is found to be 0.9.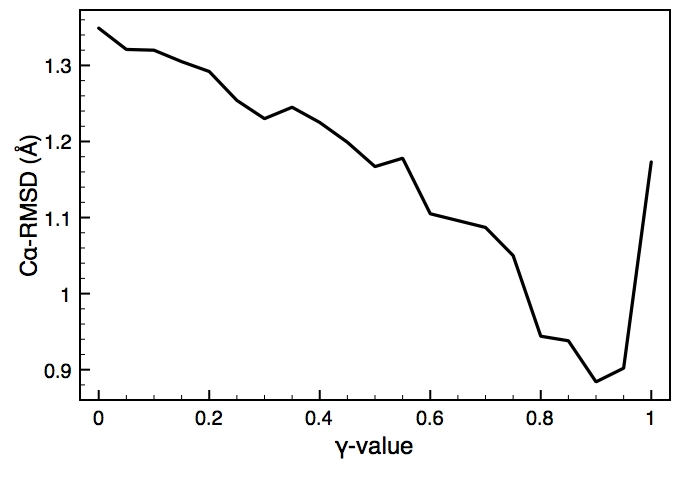
den_strength:
The strength should always be high enough to keep the local geometry in shape. A value between 0.2 and 0.4 is usually a good choice. If you feel the structure does not move enough, even for larger gamma values, you can decrease this value. However, if you have secondary structure information, it might be better to use the den_loop parameter, to decrease only the strength of restraints within loops, this is described in more detail in the EF-2 tutorial. Using a value larger than 0.5 is not recommended: the computation could become unstable.
den_upper / den_lower: The DEN restraints are chosen randomly between atoms pairs that are within the distance interval defined by these two parameters. 3 - 15 Å is the default, which should be fine in most cases.
den_resid_range: The choice of the DEN restraints can be further restricted to atom pairs that are within a given range of residue numbers, i.e. selected atom pairs will
This tutorial shows a very basic example of the usage of DireX to flexibly fit a protein structure into a low-resolution density map. The fitting of this small helix is done in just a few seconds, which allows to conveniently test the effect of the various input parameters.
A perfect α-helix (extended-helix.pdb) built with Chimera is
used as the starting structure. This starting structure
serves in this case also as the DEN reference structure
(see Introduction to DEN Method).
The target structure (target.pdb) is a kinked helix that
has a root-mean-square deviation (RMSD) of about 4.7 Å.
We first create a 10 Å density map based on this (kinked) target
structure map with DireX.
The program call can be found in the run.sh bash script:
# Make density
direx -f mkdensity.par -pdb target.pdb -cur cur_mkdensity.pdb -curmap
kinked-density.mrc -map self -map_apix 2.0
The goal is now to fit the straight extended helix into the kinked density map to obtain a fitted structure that gets as close as possible to the target structure.
The actual structure refinement is then performed with (see also the run.sh script):
# Run refinement
direx -f refine.par -pdb extended-helix.pdb -cur current.pdb
-curmap curmap.mrc -map kinked-density.mrc -ox s.xtc
IMPORTANT: DireX determines the bond lengths and angles directly from the PDB file and it does not optimize those values. It is therefore recommended that you use a (energy minimized) structure with correct bond lengths and angles.
Also, note that only lines starting with "ATOM" and "HETATM" are read, everything else is ignored.
The file current.pdb holds the current coordinates and is updated after
each iteration. After the refinement is finished it therefore
contains the final fitted structure.
Analysis
The final fitted structure has an RMSD (Ca-atoms) of 0.9 Å to the target structure. The file mapcc.dat contains the correlation between the model map and the target density map. This file can be viewed e.g. with the plot program xmgr (or grace).
xmgr mapcc.dat
vmd -f extended-helix.pdb traj.xtc -m kinked-density.mrc
Optimization
An important part of finding the optimal parameters is to balance the fitting to the density map and at the same time obtain a reasonable structure in terms of local geometry, ramachandran statistics, and secondary structure. The DEN restraints are the key element in DireX to control this balance. In the next few steps, we will play with some parameters that define the DEN restraints:
den_no:
The number of restraints should be at least equal to the number of atoms, but typically a better choice would be factor of two. In this case we therefore use 412.
den_gamma:
This is one of the key parameters in the DEN approach. In the present case, the DEN reference model is the same as the starting model (extended-helix.pdb). A small γ-value will keep the structure close to this reference model, and a large γ-value will allow larger deformations of the network and therefore should result in higher map correlations. There is usually an optimum choice of den_gamma that best balances reference information and density fit. In principle, this optimum value should be determined by cross-validation (this is not yet part of the tutorials). Since we know the correct target structure for this case, we will compute the RMSD (root mean square deviation) to the target structure to assess the effect of the γ-value. The three plots on the right show the RMSD, the fraction of residues that are within the allowed region of the Ramchandran plot, and the density map correlation. From the RMSD plot, the optimal γ-value is found to be 0.9.
den_strength:
The strength should always be high enough to keep the local geometry in shape. A value between 0.2 and 0.4 is usually a good choice. If you feel the structure does not move enough, even for larger gamma values, you can decrease this value. However, if you have secondary structure information, it might be better to use the den_loop parameter, to decrease only the strength of restraints within loops, this is described in more detail in the EF-2 tutorial. Using a value larger than 0.5 is not recommended: the computation could become unstable.
den_upper / den_lower: The DEN restraints are chosen randomly between atoms pairs that are within the distance interval defined by these two parameters. 3 - 15 Å is the default, which should be fine in most cases.
den_resid_range: The choice of the DEN restraints can be further restricted to atom pairs that are within a given range of residue numbers, i.e. selected atom pairs will