
|
DireX - Tutorial Kinked Helix at High Resolution |
In this tutorial the focus is on sidechain placement. The starting structure is the same extended helix as in the previous low-resolution example. This test case is constructed such that the sidechains can be in significantly different orientations than in the starting structure.
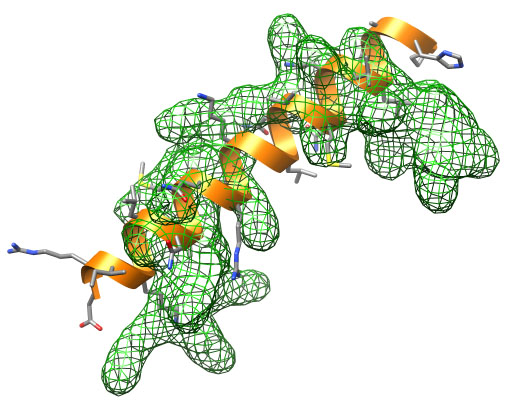 Initial structure (orange) and target density (green).
Initial structure (orange) and target density (green).
Clearly a 3 Å density map (and this ideal map in particular) defines the structure and also most sidechains quite well. We therefore switch off the DEN restraints on the sidechains which is achieved by setting den_sidechain to zero. This parameter defines a factor that is multiplied to the strength of those DEN restraints that involve sidechain atoms.
You will immediately realize that running this case takes longer than the low-resolution case. There are two reasons: (1) density map has more grid points which makes the computation slightly more costly and (2) the sampling takes longer since the 'energy landscape' (the fit function) is more rugged and has many more local minima that need to be crossed. Finding the correct optimum becomes more difficult. In some cases it might even be advisable to filter the density map to lower a resolution in a first round of refinement, and then increase the resolution in subsequent rounds until the highest resolution is reached.
The refinement is performed here in two steps: The first DireX run samples more aggressively to cross 'energy' barriers and the second run is more like a minimization (although not in a strict sense, remember that DireX does not use gradients to move atoms but instead uses a geometry-based sampling algorithm). The first DireX run:
# Run refinement
direx -f refine.par -pdb extended-helix.pdb -cur current-refine.pdb -curmap curmap.mrc -map kinked-density.mrc -ox traj.xtc
# Run minimization
direx -f min.par -pdb extended-helix.pdb -p current-refine.pdb -cur current-min.pdb -curmap curmap-min.mrc -map kinked-density.mrc -ox traj-min.xtc
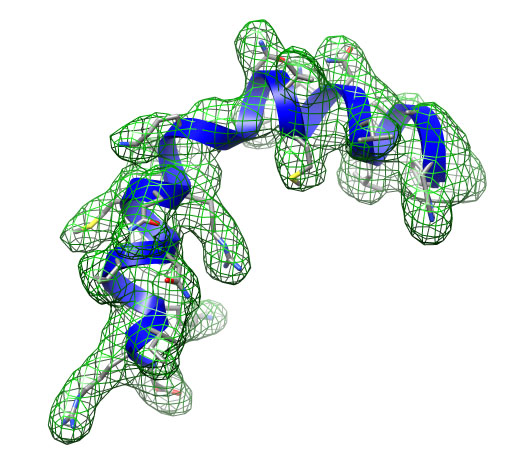 Fitted structure (blue) and target density (green grid).
Fitted structure (blue) and target density (green grid).
|
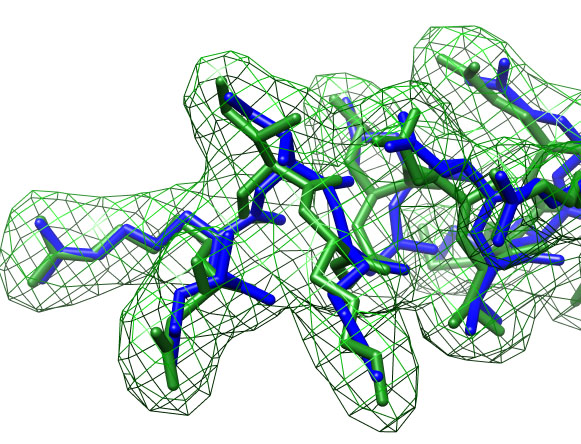 Close-up view of fitted structure (blue), target (correct) structure (green) and target density (green grid).
Close-up view of fitted structure (blue), target (correct) structure (green) and target density (green grid).
|
Analysis
Look at the resulting trajectories with VMD:
vmd -f extended-helix.pdb traj.xtc traj-min.xtc -m kinked-density.mrc target.pdb
Reading the target structure as well helps to evaluate the final fitted structure. You might also want to read in curmap.mrc to compare it to the target map kinked-density.mrc.As an exercise, you can switch off the DEN restraints (set use_den = no in refine.par and min.par). You will find that the DEN restraints will help to guide the refinement towards the optimal structure even at this (relatively) high resolution.